
Mapování genomů
Mapování genomu určitého organizmu je složitý proces. Pro co nejpřesnější představu o genové mapě zkoumaného organismu musíme zjistit následující fakta:
- Kolik má organismus chromozomů
- Na kterém chromozomu se jaký gen nachází
- V jakém pořadí jsou geny na chromozomu umístěny; jak jsou od sebe geny vzdáleny
V současné době je již běžné i sekvenování genomů, což je proces, během kterého zjistíme kompletní sekvenci nukleotidů jaderné molekuly DNA organismu.
Mapování genomu
Počet chromozomů lze stanovit běžnými mikroskopickými technikami pomocí vhodného cytogenetického barvení. Složitější již je umisťování genů na jednotlivé chromozomy. Za tímto účelem byly vyvinuty speciální metody (hybridomová metoda), kdy se vytvářely buněčné kultury s kombinovanou chromozomovou výbavou (například člověka a myši). V těchto kulturách se postupně eliminovaly lidské chromozomy (nebo jejich části) a vyhodnocovala se přítomnost produktu sledovaného v závislosti na zbylých chromozomech (pokud v buňce zbyl pouze jeden lidský chromozom a produkt hledané genu se stále tvořil, potom bylo možné tento gen lokalizovat na onen chromozom). Tato metoda byla velmi zdlouhavá a vyžadovala velikou zkušenost.
Známe-li proteinový produkt genu, můžeme zkusit z primární struktury - tedy pořadí aminokyselin - sestavit sekvenci nukleotidů, které by toto pořadí kódovaly. Nyní můžeme zkusit, zda takto vytvořené sondy budou hybridizovat s nějakou sekvencí na některém chromozomu. Využíváme při tom metody in situ hybridizace, kdy necháváme hybridizaci proběhnou takříkajíc "na místě" - tedy například přímo na podložním sklíčku mikroskopu, ve kterém můžeme rovnou sledovat, zda na některém chromozomu značená (např. fluorescenčním barvivem) sonda hybridizuje.
Pro stanovení pořadí a vzdáleností genů na chromozomech se využívá zákonů genové vazby a tříbodového pokusu.
Metody sekvenování
Pro určení přesné sekvence nukleotidů v úseku DNA byly vynalezeny dvě metody - Sangerova a Maxam & Gilbertova. Popíšeme si princip Sangerovy metody, neboť je více používaná.
Sangerova metoda využívá speciální vlastnosti speciálních nukleotidů - 2', 3' dideoxyribonukleotidtrifosfátů. Tyto nukleotidy (ddATP, ddCTP, ddGTP a ddTTP) nemají na 3' uhlíku ribosy OH skupinu, z čehož plyne, že na tento konec již nemůže být žádný další nukleotid navázán.
Nyní si představte, že máme v reakční směsi namnoženou jednovláknovou DNA (jejíž sekvenci chceme znát), DNA polymerasu, příslušné primery (aby DNA polymerasa mohla začít pracovat), dostatek deoxyribonukleotidtrifosfátů (dATP, dCTP, dGTP a dTTP) pro syntézu a ještě určité množství jednoho typu dideoxyribonukleotidtrifosfátu - dejme tomu ddATP. Co se stane - polymerasa začne od nasednuvších primerů doplňovat sekvenci druhého vlákna. Pokaždé, když polymerasa doplňuje dATP do řetězce, je určitá pravděpodobnost, že namísto dATP použije ddATP. Pokud je zařazen ddATP, potom polymerace na tomto místě končí. Necháme-li tedy takovouto reakci proběhnout, získáme velké množství různě dlouhých oligonukleotidů, které budou všechny končit adeninem (dojde k zastavení polymerace u ddATP).
Pokud necháme proběhnout stejnou reakci, tentokrát s ddCTP, ddGTP a nakonec i ddTTP, dostaneme 4 směsi oligonukleotidů, přičemž v každé směsi budou oligonukleotidy končit příslušnou bází.
Klasická metoda vyhodnocení spočívá v provedení elektroforézy, přičemž jsou v gelu vytvořeny 4 dráhy, každá pro jinou oligonukleotidovou směs.
Obrázek 1 - Výsledek 4-dráhové elektroforézy
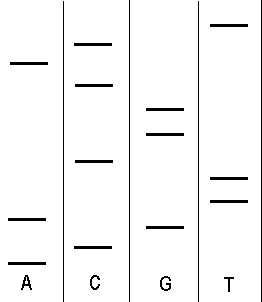
Všimněme si, že díky rozdílné délce vzniklých oligonukleotidů, doputuje každý z nich rozdílně daleko. Budeme vyhodnocovat od spodu (od nejkratšího oligonukleotidu, který doputoval nejdál - pokud je start nahoře). Všimněme si, že nejníže je oligonukleotid končící adeninem. O něco výše je oligonukleotid končící guaninem. Tímto způsobem můžeme přečíst celou sekvenci:
5' ACGATTCGGCACT 3'
V současné době existují moderní přístroje - sekvenátory - ve kterých se využívá dideoxyribonukleotidů, které jsou značeny různými fluorescenčními barvivy (pro každou bázi je jiná barva). Výslednou sekvenci pak přístroj určuje na základě tohoto barevného značení.
Maxam & Gilbertova metoda se vyhodnocuje podobným způsobem (elektroforeticky), ovšem reakce, při které vznikají různě dlouhé oligonukleotidy, nevyužívá polymerace podle vzorového vlákna, ale specifického chemického štěpení DNA za určitými nukleotidy.
Obrázek 2 - Výsledek moderní sekvenace úseku DNA

Výsledky mapování
Výsledky zkoumání genomu jsou různé (výsledky nejznámějšího projektu - přečtení lidského genomu - najdete v kapitole Genetika člověka.). Existují genové mapy, které jsou vytvořeny na základě výpočtů, vycházejících ze zákonů genové vazby (vzdálenosti v centiMorganech - cM). Existují také fyzické mapy, založené na přesné pozici genu v sekvenci DNA (vzdálenosti v párech bází - pb). Znalost kompletní sekvence genomu je nesmírně užitečná, sama však pro přesnou lokalizaci všech genů nestačí. Velké množství genů (přesněji jejich mutovaných forem) bylo objeveno až jako původci různých dědičných onemocnění. I obecně platí, že nejlépe funkci genu poznáme, když mutací tento gen vyřadíme z funkce.
Vzhledem k tomu, že z etických důvodů nelze provádět cílenou mutagenezi a některé další pokusy na člověku, jsou pro další výzkum lidského genomu nedocenitelné výsledky získané u jiných organismů, které jsou potom porovnávány s dosavadními výsledky výzkumu u člověka (komparativní genomika).
Obrázek 3 - Chromozomová mapa s vyznačením pozic genů HLA systému na lidském chromozomu 6





